



AAA family proteins are involved in a variety of cellular processes including protein degradation, membrane biogenesis and fusion, and reconstitution of organelles (Fig. 1). They form ring-shaped oligomers. Upon ATP hydrolysis, they unfold and translocate substrate proteins or disassemble protein complexes. Thus, they can be thought of as novel types of molecular chaperones. We are investigating the cellular functions and molecular mechanisms of various AAA proteins. Recent studies have implicated AAA proteins in a number of human genetic diseases including neurodegenerative disorders.
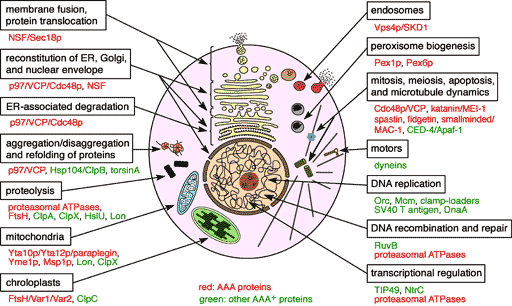
Fig. 1.Various cellular functions of AAA family proteins.
Common structure and diverse function of AAA family proteins
AAA family proteins are a subfamily of the Walker-type NTPases. Their functional architecture is a hexameric ring with a central pore (Fig. 2). Recent studies have indicated that they represent a novel class of molecular chaperones, which unfold proteins and disassemble protein complexes. They are highly conserved from prokaryotes to humans. Interestingly, the number of AAA members and their functional distribution is similar in eukaryotes as diverse as yeast, worms, flies and humans. We are interested in elucidating whether there is a common molecular mechanism underlying the diverse functions of the family members.
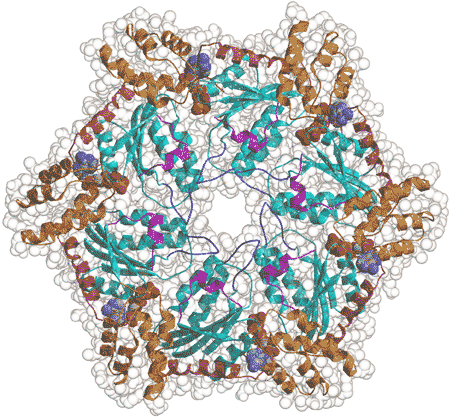
Fig. 2. Hexameric ring structure of the AAA family ATPase.
1. Common molecular basis of the AAA family of ATPases
AAA family ATPases possess a family-specific motif, the SRH (second region of homology), in addition to Walker A and B motifs. We have carried out an alanine-scanning mutagenesis analysis of the SRH, and have collaborated with A. J. Wilkinson (Univ. of York) to solve the crystal structure of the ATPase domain of the Escherichia coli AAA protease FtsH (Fig. 3). These analyses have led us to propose an inter-subunit catalysis mechanism for ATP hydrolysis by the AAA ATPase (Fig. 4). In vitro studies using mutants of the Caenorhabditis elegans fidgetin homologue FIGL-1 have provided direct evidence for the model. It has also been suggested that a conformational change upon ATP binding is required for proper orientation of the arginine finger. The mode and the kinetics of ATP binding is being visualized using total internal reflection fluorescence microscopy as a collaboration with T. Nishizaka (Gakushuin Univ.). In the hexameric ring, there is a conserved aromatic residue, which lines the central pore (Fig. 5). Functional assays have indicated that bulkier uncharged/apolar residues at this position are essential for retention of activity, suggesting a role for this residue in threading unfolded polypeptides through the enzyme’s central pore. We have investigated degradation of native and engineered substrate proteins. Katanin and spastin are AAA proteins, which possess microtubule-severing activity. Mechanochemical analysis of purified katanin and spastin is being carried out. Dynamic movements of AAA proteins and their complexes with substrates are also being analyzed by high speed atomic force microscopy as part of a collaboration with T. Ando (Kanazawa Univ.).
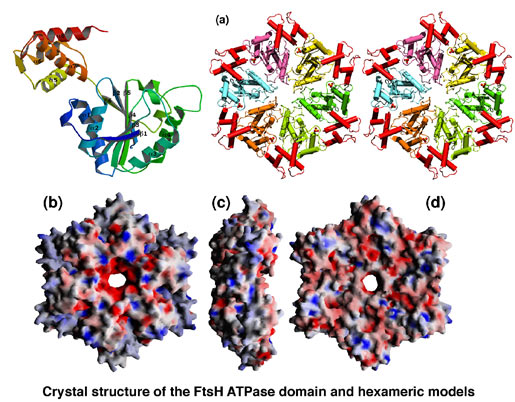
Fig. 3. Crystal structure of the ATPase domain of FtsH.
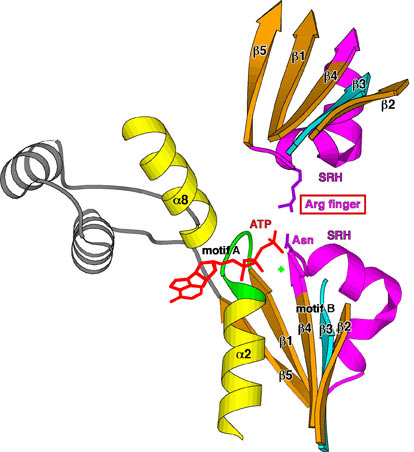
Fig. 4. Inter-subunit catalysis model of ATP hydrolysis
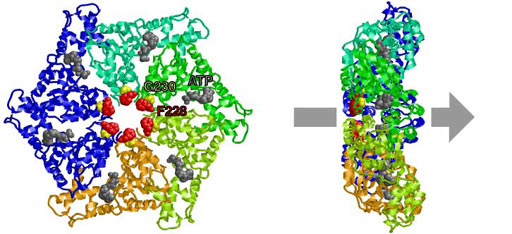
Fig. 5. Conserved residues at the central pore.
2. Cellular functions of the AAA protease, FtsH
E. coli FtsH is an essential membrane-bound AAA protease (Fig. 6). FtsH degrades the heat shock transcription factor, s32, thus regulating the heat shock response. LpxC is the committed enzyme involved in the biosynthesis of lipopolysaccharide (LPS), a major component of the outer membrane. FtsH degrades LpxC and down-regulates LPS synthesis thus maintaining a balance in the biosynthesis of phospholipids and LPS. Genetic analysis has indicated that regulated proteolysis of LpxC by FtsH is essential for cell viability. As FtsH also degrades abnormal membrane proteins, FtsH is an important regulator of membrane formation and quality control. In ftsH mutants, partitioning of mini-F plasmids is disturbed.
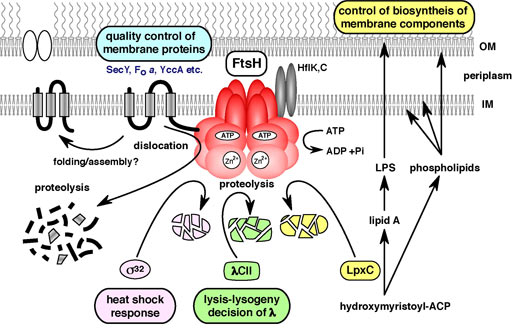
Fig. 6. Cellular functions of FtsH.
Cellular functions of AAA proteins in C. elegans
C. elegans possesses 24 AAA protein family members (Fig. 7). Some of these proteins are homologous to those involved in human genetic diseases, such as Pex1p and Pex6p in peroxisome diseases, paraplegin and spastin in hereditary spastic paraplegia (HSP), and p97/VCP in inclusion body myopathy associated with Paget disease of the bone and frontotemporal dementia (IBMPFD) (Fig. 8). We have studied the molecular mechanisms and cellular functions of C. elegans AAA family proteins, especially those related to human genetic diseases.
Paraplegin homologs (SPG-7 and PPGN-1): These are homologs of bacterial FtsH, and they are AAA proteases localized in the mitochondria (Fig. 9). Their expression patterns are distinct. The effects of RNAi on the two genes are also different; a mixed phenotype (larval lethal and slow growth) results with SPG-7, while no significant phenotype is associated with silencing of PPGN-1. Histochemical assays and EM observations (by T. Suzaki; Kobe Univ.) of spg-7(RNAi) suggest there are mitochondrial defects. Spastin and fidgetin homologs (SPAS-1 and FIGL-1): Spastin is a second AAA protein involved in HSP. Spastin is related to microtubule dynamics as described above. Another AAA protein, fidgetin, which is closely related to spastin, has been identified as a causative factor of the ‘fidget’ phenotype in mice, which is characterized by reduced or absent semicircular canals and small eyes. The roles of the C. elegans homologs of these AAA proteins are being studied.
p97/VCP homologs (CDC-48.1 and CDC-48.2): p97/VCP is a multifunctional AAA protein, which is involved in the reconstitution of endoplasmic reticulum (ER), Golgi apparatus, and nuclear envelope, the ubiquitin-proteasome pathway, and ER-associated degradation (ERAD). Mutations in human p97/VCP cause IBMPFD. It is also implicated in apoptosis and polyglutamine diseases. RNAi assays of C. elegans p97/VCP homologs, CDC-48.1 and CDC-48.2, revealed that simultaneous disruption of both genes was lethal at the embryo stage. We are currently investigating the roles of p97/VCP homologs in ERAD, sex determination, and aggregation/disaggregation of polyglutamine repeats (Fig. 10).
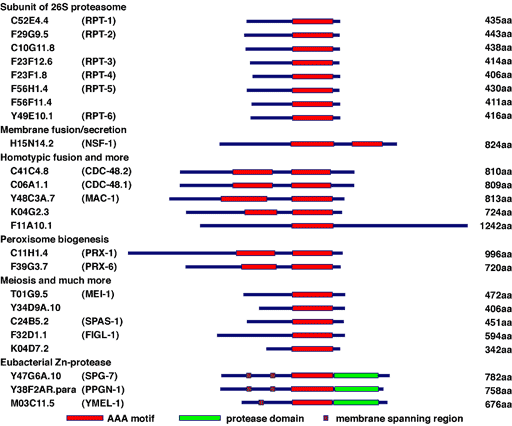
Fig. 7. AAA proteins in C. elegans.
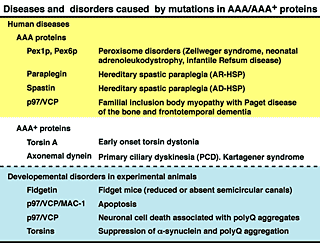
Fig. 8. Diseases and disorders caused by mutations in AAA/AAA+ proteins.
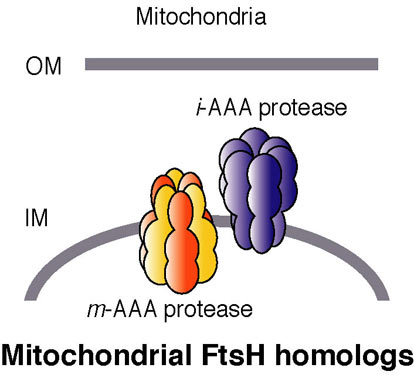
Fig. 9. AAA proteases localized in the mitochondria.
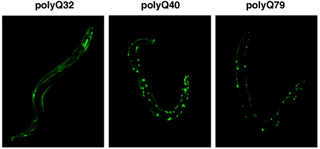
Fig. 10. Polyglutamine aggregation in C. elegans.





