









Shunsuke Tanigawa, Mazharul Islam, Sazia Sharmin, Hidekazu Naganuma, Yasuhiro Yoshimura, Fahim Haque, Takumi Era, Hitoshi Nakazato, Koichi Nakanishi, Tetsushi Sakuma, Takashi Yamamoto, Hidetake Kurihara, Atsuhiro Taguchi, and Ryuichi Nishinakamura. Organoids from nephrotic disease-derived iPSCs identify impaired NEPHRIN localization and slit diaphragm formation in kidney podocytes
Stem Cell Reports on line Aug 30, 2018
Mutations in the NPHS1 gene, which encodes NEPHRIN, cause congenital nephrotic syndrome, resulting from impaired slit diaphragm (SD) formation in glomerular podocytes. However, methods for SD reconstitution have been unavailable, thereby limiting studies in the field. In the present study, we established human induced pluripotent stem cells (iPSCs) from a patient with an NPHS1 missense mutation and reproduced the SD formation process using iPSC-derived kidney organoids. The mutant NEPHRIN failed to become localized on the cell surface for pre-SD domain formation in the induced podocytes. Upon transplantation, the mutant podocytes developed foot processes, but exhibited impaired SD formation. Genetic correction of the single amino acid mutation restored NEPHRIN localization and phosphorylation, colocalization of other SD-associated proteins, and SD formation. Thus, these kidney organoids from patient-derived iPSCs identified SD abnormalities in the podocytes at the initial phase of congenital nephrotic disease.
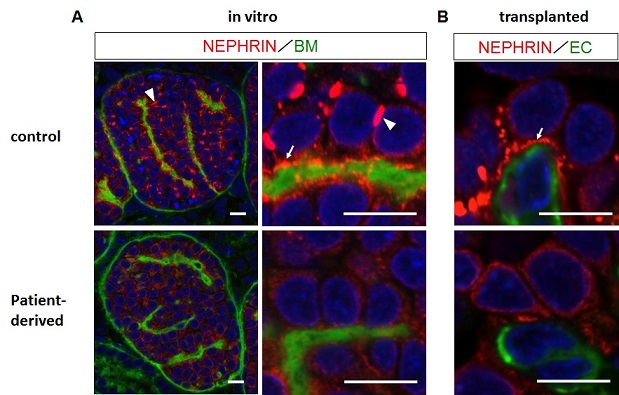
Figure.Impaired localization of NEPHRIN in the patient-derived glomerular podocytes
A: Glomerular podocytes induced in vitro from iPS cells
B: Glomerular podocytes generated upon transplantation
NEPHRIN+ pre-SD domains, which are formed in the lateral (arrowheads) and basal (arrows) regions of the control podocytes, are not detectable in the patient-derived podocytes.
BM: basement membrane; EC: endothelial cells
Scale bars:10 µm


