








1. Recruitment of Rad6-Rad18-Polη complex to stalled replication fork.
Cellular DNA is continually damaged by intrinsic sources; reactive oxygen species and extrinsic sources; UV light irradiation. If these DNA damages are not repaired, accumulation of the DNA damages cause cellular death or carcinogenesis. Most DNA lesions are removed prior to DNA replication by the nucleotide excision and base excision repair pathways. However, the nucleotide excision repair pathway is not always efficient to repair the DNA lesions; cyclobutane pyrimidine dimer. Therefore, unrepaired lesions encountered by the DNA replication machinery during S-phase cause replication fork stalling. Cell death may occur unless DNA synthesis resumes at stalled replication forks. This resumption process is operationally defined as post-replication repair (PRR), and is characterized by re-initiation of DNA replication without removal of the lesion on a template strand (1). PRR is observed in diverse species from E. coli to humans and is hypothesized to involve three major steps: translesion DNA synthesis (TLS), recombination and template switching. The term, PRR has been used interchangeably with “DNA damage tolerance” or “DNA damage avoidance”.
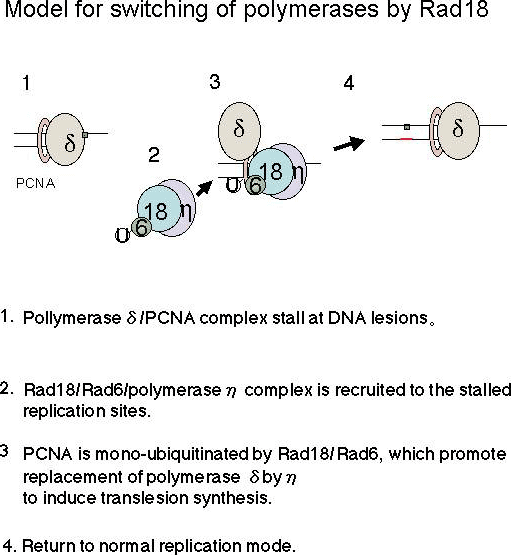
Fig. 1.
In the budding yeast S. cerevisiae, genes belonging to the RAD6 epistasis group are involved in the PRR pathway, where RAD6 and RAD18 (coding for ubiquitin-conjugating enzyme E2 and ubiquitin ligase E3, respectively) play a pivotal role. In vertebrate cells, we have identified a homologue of Rad18 (2), while two homologues of Rad6 were identified (designated Rad6A and Rad6B). PCNA was shown to be mono-ubiquitinated in a Rad6/Rad18 dependent manner, which is necessary for tolerance of DNA damage (3). In mammalian cells, translesion synthesis DNA polymerases operate to circumvent replication blocks. Among TLS polymerases mostly belonging to the Y-family, Polη is of great interest because the gene encoding Polη is mutated in a cancer-prone hereditary disorder, xeroderma pigmentosum variant (XPV) (4, 5).
We and other researchers have revealed that Polη is thought to replace replicative polymerase δ at stalled replication forks through interactions with Rad18 and mono-ubiquitinated PCNA (6, 7). We have proposed the model how Rad18 guide Polη to stalled replication sites and promote replacement of replicative polymerase by TLS polymerase (refer to the Figure). In UV-irradiated human cells, Polη and Rad18 are recruited to nuclear foci in a UV-dose and time dependent manner (6). Polη/Rad18 foci are colocalized with PCNA, suggesting that these are sites of stalled replication. A mutant Polη devoid of focus-forming activity cannot complement the XPV defect (8), demonstrating the importance of Polη/Rad18 recruitment to stalled replication forks for initiation of translesion synthesis. Despite the importance of Polη and Rad18, the molecular mechanism by which they are recruited to stalled replication forks are not well understood.
2. Elucidation of the role of Rad18 in stabilization of genome.
We have prepared mouse Rad18-knockout cells (9). These cells had almost the same growth rate as wild-type cells and manifested phenotypes: hypersensitivity to multiple DNA damaging agents and a defect in postreplication repair. In the knockout cells, spontaneous sister chromatid exchange (SCE) occurred with twice the frequency observed in normal cells. After mild DNA damage, SCE was three fold higher in the knockout cells, while no increase was observed in normal cells. Stable transformation efficiencies were ~20-fold higher in knockout cells, and gene targeting occurred with ~40 fold higher frequency than in wild-type cells at the Oct3/4 locus. These results indicate that dysfunction of Rad18 greatly increases both the frequency of homologous as well as illegitimate recombination, and that RAD18 contributes to maintenance of genomic stability through postreplication repair. In S. cerevisiae, Rad18 is also known to contribute to maintenance of genomic stability to prevent gross chromosomal rearrangements(10). Therefore, this role of Rad18 is conserved from yeast to humans.
References
- Broomfield, S., T. Hryciw, and W. Xiao. 2001. DNA postreplication repair and mutagenesis in Saccharomyces cerevisiae. Mutat Res 486:167-84.
- Tateishi, S., Y. Sakuraba, S. Masuyama, H. Inoue, and M. Yamaizumi. 2000. Dysfunction of human Rad18 results in defective postreplication repair and hypersensitivity to multiple mutagens. Proc Natl Acad Sci U S A 97:7927-32.
- Hoege, C., B. Pfander, G. L. Moldovan, G. Pyrowolakis, and S. Jentsch. 2002. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature 419:135-41.
- Masutani, C., R. Kusumoto, A. Yamada, N. Dohmae, M. Yokoi, M. Yuasa, M. Araki, S. Iwai, K. Takio, and F. Hanaoka. 1999. The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase η. Nature 399:700-4.
- Johnson, R. E., C. M. Kondratick, S. Prakash, and L. Prakash. 1999. hRAD30 mutations in the variant form of xeroderma pigmentosum. Science 285:263-5.
- Watanabe, K., S. Tateishi, M. Kawasuji, T. Tsurimoto, H. Inoue, and M. Yamaizumi. 2004. Rad18 guides polη to replication stalling sites through physical interaction and PCNA monoubiquitination. Embo J. 23:3886-96.
- Kannouche, P. L., J. Wing, A. R. Lehmann. 2004. Interaction of human DNA polymerase η with monoubiquitinated PCNA: a possible mechanism for the polymerase switch in response to DNA damage. Mol Cell 14:491-500.
- Kannouche, P., B. C. Broughton, M. Volker, F. Hanaoka, L. H. Mullenders, and A. R. Lehmann. 2001. Domain structure, localization, and function of DNA polymerase η, defective in xeroderma pigmentosum variant cells. Genes Dev 15:158-72.
- Tateishi, S., Niwa, H., Miyazaki, J., Fujimoto, S., Inoue, H., and Yamaizumi, M. 2003. Enhanced genomic instability and defective postreplication repair in RAD18 knockout mouse embryonic stem cells. Mol. Cell. Biol., 23:474-481.
- Smith, S., Hwang, J., Banerjee, S., Majeed, A., Gupta, A. and Myung, K. 2004. Mutator genes for suppression of gross chromosomal rearrangements identified by a genome-wide screening in Sacchromyces cerevisiae. Proc. Natl. Acad. Sci. USA, 101:9039-9044.









