(1) Studying for intractable diseases
We have been collecting skin-derived fibroblasts and blood cells from intractable disease with the consent of patients and establishing the disease-derived iPS cells. In disease research, we are studying congenital metabolic disorders, age-related neurodegenerative diseases and autoimmune diseases. Congenital metabolic disorders are a general term for tissue and organ disorders caused by abnormalities in the carbohydrate metabolism, lipid metabolism, or amino acid metabolism pathways. They are mainly caused by defect of enzyme activity due to the gene mutations. Main symptom includes the central nervous system disorders, and the severity of them determines the prognosis. We are mainly studying lipid metabolic disorders such as GM1 gangliosidosis (GM1G), Tay-Sach disease (TSD), sialidosis (SD), Gaucher disease (GD), and Niemann-Pick disease type C (NPC) (Figure 1).
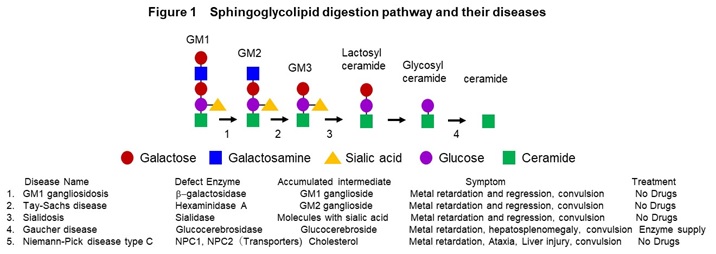
In these diseases, intermediate metabolic substances that are substrates for defective enzymes and transporters accumulate in the organs such as brain and liver. The frequency is 1-2 people per 100,000, respectively, and it is estimated that there are more than 500 patients in Japan. In addition, we are conducting research on Alzheimer’s disease, an age-related neurodegenerative disease, and scleroderma, an autoimmune disease. We have established a method to induce mature neurons that form synapses from iPS cells, and a method to analyze synapse function by imaging (Figure 2).
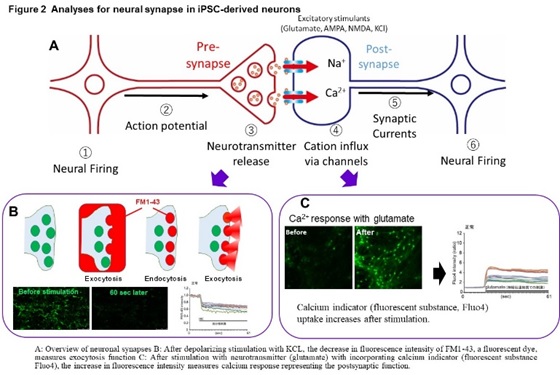
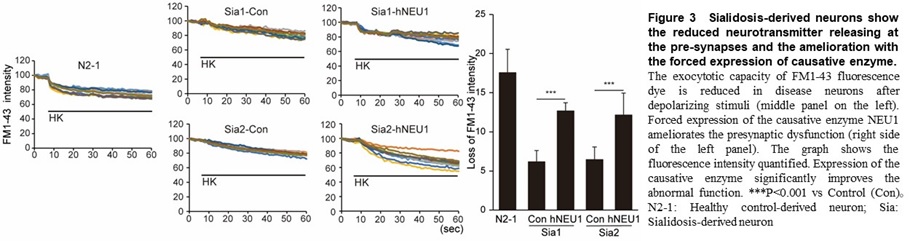
Using this method, we found a common abnormal function including a marked decrease in exocytosis (which suggests an extreme decrease in the ability to release neurotransmitters at the pre-synapse) in GM1G, TSD, and SD (Figure 3).
This phenotype was also observed in primary neurons obtained from model mice. In addition, we also found an abnormal function including an excessive response of calcium dynamics at the post-synapse in SD. In GM1G, these phenotypes can be reproduced by accumulating GM1 ganglioside, the substrate of the causative enzyme in normal neurons, suggesting the accumulation of intermediate is the cause of this abnormal function and cell death. We also identified the compounds that have the effect of suppressing cell death and improving the synaptic abnormalities with reducing accumulated GM1 ganglioside, in GM1G (Figure 4).
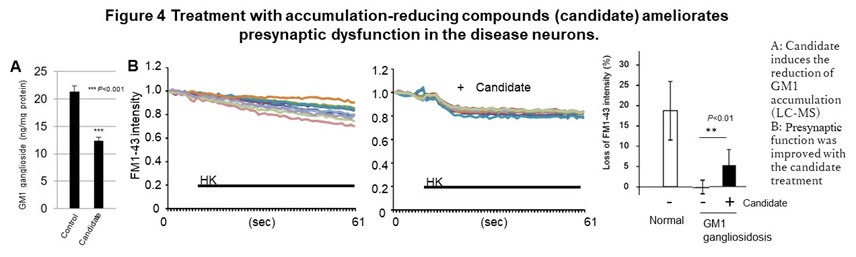
In addition, in the area of therapeutic drug development, eight drugs are currently being developed for the diseases described above, including three candidate drugs that are effective in model cells, three drugs whose efficacy has been confirmed in model animals, and two candidates under the clinical trials. In the case of congenital metabolic disorders, a compound screening system (HTS) in which accumulated substrates are visualized was established for multiple diseases based on the new concept that suppressing the accumulation of intermediate metabolites provides the therapeutic effects. Using these HTS and abnormal phenotypes in neurons to determine the efficacy, several hit chemicals were identified from a library of known drugs. These hit chemicals significantly reduced the accumulation of GM1 ganglioside in neurons and improved the abnormal synaptic function in vitro, Furthermore, in NPC, dextrin derivatives was shown to suppress the progression of motor dysfunction caused by neuropathy in disease model mice and prolonging survival time.
(2) Studying for gene repair
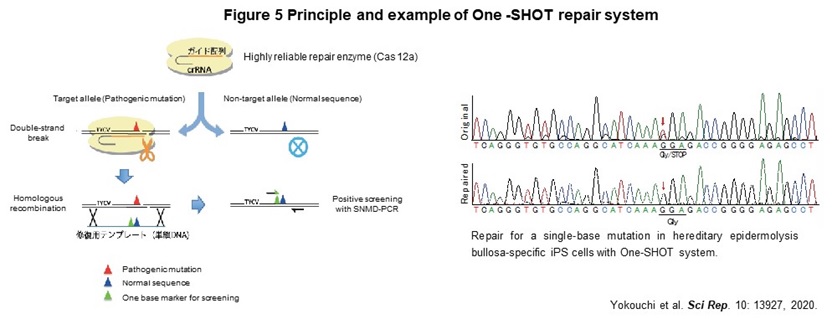
Human genome contains about the 40,000 disease-specific single nucleotide variants (SNVs), which cause the genetic diseases. As a first step toward the realization of genome repair medicine for SNVs, we developed One-SHOT, an allele-specific single nucleotide substitution method in disease-specific iPS cells (Figure 5). While previous methods such as CORRECT and MhAX required two rounds of genetic manipulation, this method can be performed and completed with 1. a single genetic manipulation, 2. development of a new single-base mismatch detection PCR method to identify edited clones, and 3. transgenic clones with temporary expression of drug resistance genes. In addition, combining 4. enrichment of a new editing tool with high target discrimination ability to identify single nucleotide mutations, 5. labor-saving culture operation by maintaining a master plate, and 6. use of ssODN as a template, allows us to reduce the time and cost to about 1/3 of those of previous ones.
(3) Studying for mesenchymal stem cells
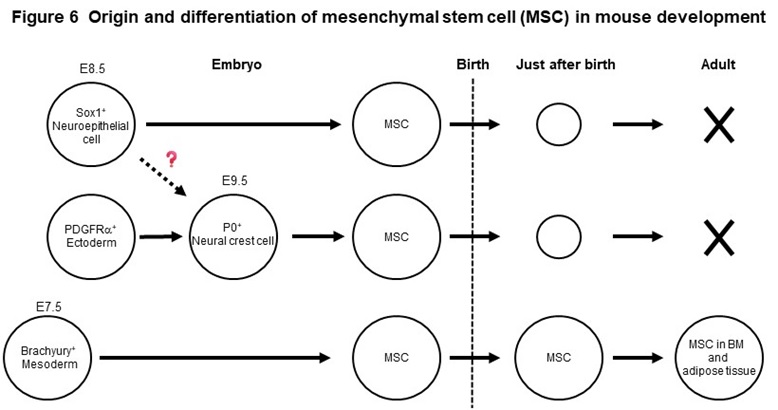
Mesenchymal stem cells (MSCs) are widely distributed in our body, such as bone marrow and adipose tissue and exhibit a fibroblast-like morphology. MSCs, that can easily proliferate in vitro, have the ability to differentiate into bone, cartilage, and adipocytes, as well as various other mesenchymal cells such as muscle cells. Using genetically modified mice, we found that MSCs present in adult bone marrow and adipose tissue originate from the fetal mesoderm (Figure 6). We have also shown that MSCs born from neuroepithelial cells and neural crest cells exist in the embryonic period, but disappear rapidly after birth (Figure 6). Based on these new findings, we have succeeded in inducing two types of MSCs with different origins, mesodermal and neuroepithelial-derived MSCs, from human iPS cells. The two MSCs have therapeutic efficacy on pressure ulcers and osteoarthritis model mice, but the effects are different. We revealed that one of the reasons for this is the difference in secretory factors produced by the two stem cells.