









私達の体を構成している細胞内の遺伝子の本体である DNAは、常に傷害を受けています。傷害された細胞では、細胞周期の進行が抑制され、遺伝子の損傷はDNA修復システムにより修復されます。このようにして、貴重な遺伝情報は正確に保たれています。細胞周期調節機構またはDNA修復機構に異常が発生すると、遺伝子に変異が蓄積し、発癌や老化の原因となります。細胞周期調節機構およびDNA修復機構を研究することを通して、細胞が癌化するのを防御し癌の治療に応用すること、また再生医学を推進するために「幹細胞の維持機構」を解明することをめざしています。
1 . 損傷トレランスとは?
紫外線の照射、放射線または活性酸素など、さまざまな原因により、遺伝子物質である DNAは傷害されています。DNAに形成された損傷は、ヌクレオチド除去修復(nucleotide excision repair : NER)などの修復機構により修復されます。修復されたDNAは複製酵素により複製され、細胞は増殖してゆきます (図1 上段)。しかし、細胞に備わっている DNA 修復能力は完全ではないため、 DNA 複製時に鋳型鎖に損傷が残存していると、 DNA複製が停止し細胞増殖が停止してしまうことがあります。 この危機的状況を回避するシステムが「損傷トレランス」であり、損傷を含む DNA 鋳型 鎖を複製するためのシステムです (図1 下段)。損傷トレランスは、損傷乗り越え複製およびテンプレートスイッチ(鋳型鎖乗り換え)の二つの機構から構成されます(図1 下段)。 損傷乗り越え複製は、損傷乗り越え複製酵素と呼ばれる特殊な複製酵素により損傷を含むDNA鋳型鎖も複製されるシステムです。損傷の種類およびそれに対処する損傷乗越え複製酵素の種類に応じて、複製エラー頻度の低いタイプと高いタイプの複製があります。損傷トレランスにより損傷部位を回避してDNAを複製した後に、Polδによる通常のDNA複製が再開されると考えられています。「損傷トレランス」は損傷を直接修復する機構ではなく、鋳型鎖に損傷が残っていても、その鋳型鎖の複製を続行して細胞増殖するための機構です。鋳型鎖に残った損傷は、DNA複製後にNERなどの修復機構により修復されると考えられています。なお、損傷トレランスは複製後修復(postreplication repair)または損傷回避(DNA damage avoidance)と呼ばれることもあります。損傷トレランス機構が破綻すると、染色体が不安定になり、発癌または細胞死の原因となるため、その機構を理解するために研究しております。
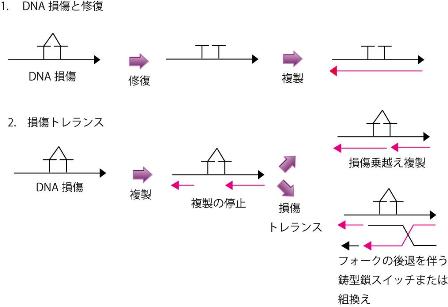
図 1. DNA 修復と損傷トレランス
( 上段 ) 細胞に UV が照射されると、 DNA にチミン 2 量体が形成される。この DNA 損傷 (DNA の傷 ) は、種々の修復システムにより修復される。修復された DNA は複製酵素により複製される。 ( 下段 ) DNA 複製時またはその直前に形成された DNA 損傷は、修復される前に DNA 複製酵素に出会う。通常の DNA 複製酵素は損傷部位を含む鋳型鎖を複製することはできないため、複製が停止してしまう。この危機的状況は「損傷トレランス」機構により回避され、複製が再開される。損傷トレランスは、損傷を修復するシステムではなく、 DNA 複製を続行させるためのシステムである。
2 .損傷乗り越え複製 (TLS)
高等真核生物では、損傷乗り越え複製が重要な役割をもつ。 1996年になるまで、通常の複製酵素であるPolδが損傷に遭遇した場合に、細胞がどのようにしてこれを乗り越えて複製してゆくのかわかっていなかった。常染色体劣性遺伝疾患である色素性乾皮症バリアント群(xeroderma pigmentosum variant)は、日光露光部での色素沈着、高頻度発がんなどを特徴とする。1999年に原因遺伝子を特定され、原因遺伝子がコードしているタンパクは、損傷を効率良く乗り越えて複製を行うDNA複製酵素であることが解明され、ヒトDNAポリメラーゼη(polymeraseη : Polη)と命名された 2) 。 Polηは、どのようにして損傷を乗り越えて複製することができるのであるのか? Polηは、鋳型鎖を複製する時に、高い頻度で誤ったヌクレオチドを挿入する性質があることがわかり、忠実度(正確性)の低い複製酵素であることが判明した 3) 。Polηを含む損傷乗越え酵素は、ヌクレオチドを認識する結合ポケットが大きいため、損傷を乗り越えてヌクレオチドを対合させて複製できる代償として、忠実度が低い性質があると考えられた。したがって、生体内で損傷のない状態ではTLS酵素が機能しないように厳密に制御されていると考えられている。
3 .RAD18によるPCNAのユビキチン化
出芽酵母を用いた遺伝学的解析により、 RAD6およびRAD18を中心とする遺伝子群(RAD6エピスタシス群)が損傷トレランスに関与しており、 TLS は RAD 18 により制御されている可能性が指摘されてきた 4) 。 RAD6およびRAD18は、それぞれ一次構造の特徴からユビキチン結合酵素(E2)およびユビキチンリガーゼ(E3)と推測されてきた。2002年に、出芽酵母のDNAに損傷が生じると、DNA複製に必要な因子の一つである増殖細胞核抗原(proliferating cell nuclear antigen : PCNA)がRAD6およびRAD18遺伝子に依存してモノユビキチン化され、この反応が損傷トレランスで重要な役割を果たしていることが示された 5) 。我々は、ヒトのRAD18遺伝子が特定し、損傷トレランスに関与していることを明らかにした 6) 。RAD18遺伝子を欠損させたマウスES細胞は、UV照射、メチル化剤、架橋化剤等に感受性を示し、また姉妹染色分体間組換え(SCE)頻度および外来のDNAをゲノムに組み込む効率が上昇していた 7) 。このため、RAD18遺伝子はゲノムの安定化の維持に貢献していると結論した。野生型マウス由来の細胞にUVを照射してDNAが損傷されると、PCNAがモノユビキチン化されるのに対して、RAD18遺伝子を欠損させたマウス由来の細胞ではこの反応が検出されなかった。このため、哺乳類細胞でもRAD18遺伝子の機能に依存してPCNAがモノユビキチン化されることがわかった 8) 。精製したヒトRAD6-RAD18タンパク質複合体は in vitro でPCNAをモノユビキチン化する活性があることを示し、RAD6-RAD18はそれぞれ、PCNAをモノユビキチン化するE2およびE3酵素であることを確定した 8) 。
4 .通常の DNA複製酵素からTLS酵素へのスイッチング
Polηはどのようにして、損傷がある場合にのみ機能するように制御されているのであろうか? 細胞にUVを照射すると、RAD18およびPolηは細胞核内のDNA複製停止部位(損傷部位)に集積する。集積がみられない変異Polηを発現する細胞は、UV照射に対して感受性を示すため、Polηの損傷部位への集積反応は重要な反応である 9) 。UV照射により、RAD18も損傷部位へ集積する。これに対して、RAD18欠損細胞ではPolηの集積反応がみられなかったことから、Polηの細胞内局在はRAD18により制御されている 8) 。RAD18とPol h は、 DNA損傷の有無に関係なく結合する性質をもつ 8, 10) 。 この相互作用の重要性を検証するため、まずRAD18におけるPol h 結合ドメインを決定した。このドメインを欠失した RAD18は、RAD18欠損細胞のUV感受性を相補する活性を失っていた。このため、RAD18とPolηの相互作用は、損傷トレランスの機能を発現するために重要である 8) 。DNAに損傷が形成されると複製が停止するため、停止した複製フォークに長い一本鎖DNA領域ができると考えられている。精製したRAD18タンパクは、100ヌクレオチド以上の長い1本鎖DNAおよびフォーク型構造をもつDNAに強く結合する性質をもつため 11) 、Polηと結合した状態で停止した複製フォークに集積すると思われる 8) 。複製停止部位においてPolηは、どのようなメカニズムによりPolδと置き変わり(ポリメラーゼスイッチング)、TLSを開始するのであろうか? PCNAはホモ三量体のリング状構造を形成し、DNA複製酵素によるDNA鎖伸長を促進するクランプとして機能する。PolηのDNA複製活性もPCNAにより促進されると報告されている。DNA損傷に応答して集積したRAD18によりPCNAがモノユビキチン化されることにより、PolηとPCNAの相互作用が増強されると仮定すると、それが原動力となり複製の場においてPolδからPolηへ置き変わる可能性がある。未修飾のPCNAとモノユビキチン化されたPCNAを用いてPolηとの親和性を比較した結果、未修飾のPCNAに比べてモノユビキチン化されたPCNAは、Polηとの親和性が高いことがわかった 8, 12) 。これに対して、PolδではそのようなPCNAに対する相互作用に差がみられなかった。このため、DNA損傷に応答して集積したRAD18によりPCNAがモノユビキチン化されると、PolηとPCNAの相互作用が増強され、それが原動力となり複製の場においてPolδからPolηへ置き変わるというモデルを提唱した(図2)。
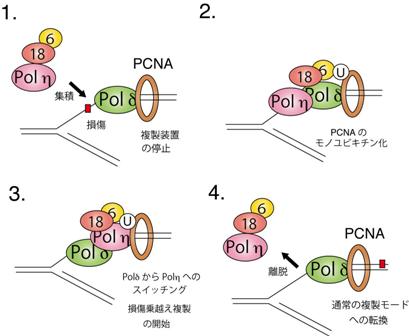
図 2. RAD18 による損傷乗越え複製の誘導モデル
(1) DNA 複製直前または複製時に DNA が傷害されると通常の複製酵素δは損傷の手前で停止してしまい、複製フォークの進行が止まる。 RAD18 は 1 本鎖 DNA を含むフォーク型 DNA に強く結合し、また損傷乗越え複製酵素ηと結合する性質をもつため、ηとともに停止した複製フォークに集積する。
(2) RAD18 は RAD6 とともに、 PCNA をモノユビキチン化する。
(3) モノユビキチン化された PCNA は、複製酵素δよりもηに高い親和性を示すため、複製部位においてδからηへの交換反応が起こる。複製部位にリクルートされたηにより、損傷を乗越えて複製が再開される ( 損傷乗越え複製 ) 。
(4) この後、δによる通常の複製モードに戻ると考えられる。
5. Yファミリ-に属するTLS酵素
TLS酵素は、Polηの他にPol i 、 Pol k および Rev1があり、これらはYファミリ-に属するTLS酵素に分類される 13) 。UBMまたはUBZと呼ばれるユビキチン結合モチーフがあるため、ユビキチン化されたPCNAに対して高い親和性を示す。DNA損傷には、CPDの他にさまざまな種類の損傷があるが、Yファミリ-に属するTLS酵素群は損傷の種類に応じて使い分けられている可能性が高い。一例として、Polηが効率良くCPDを乗り越えて複製するのに対して、Pol k はタバコに含まれるベンツパイレンにより損傷された DNAを複製する。Yファミリ-に属するTLS酵素は、ユビキチン化されたPCNAと相互作用するが、RAD18と相互作用することが知られているTLS酵素はPolηのみである。また、Rev1はPolη、Pol i および Pol k と相互作用する。 Yファミリ-に属するTLS酵素がどのようにして、種々の損傷に対応してTLSを行うのかよくわかっていないが、RAD18が損傷により停止した複製フォークを認識して結合し、PolηがRAD18とともに複製の場にリクルートされ、その後にRev1を介してPol i または Pol k が損傷の種類に応じて選択されることも予想される。
6. 精巣幹細胞における RAD18の機能
各臓器での RAD18の発現量を比較すると、精巣で高い発現がみられる。精巣の精原細胞、精母細胞などの生殖細胞で発現していることがわかった。生体でのRAD18の役割を調べるために、我々はRAD18ノックアウトマウスを作成した。RAD18ノックアウトマウスは正常に誕生し、ほぼ正常に育った。若年齢の雄マウスの生殖能力は正常であったが、加齢に伴い減少することがわかった 15) 。精巣重量も減少し、精巣の精細管内の生殖細胞が加齢に伴い欠損し、空洞化している精細管が多くみられた。このため、RAD18は長期にわたる精子形成の維持に必要であると結論した 15) 。また精原細胞のみを欠失した精細管が多くみられたことから、RAD18の欠損により損傷トレランスが不能になった精子幹細胞の自律的複製能力が低下することにより、幹細胞数が減少するため生殖細胞が枯渇すると考察した。今回、RAD18遺伝子が長期にわたる生殖細胞の維持に必要であったことから、損傷トレランス機構が生殖幹細胞の維持に必要であることが強く示唆されたので、これを明らかにすることも今後の課題である。
近年、 DNA修復機構が、生殖幹細胞または造血幹細胞の維持に重要な役割を果たしていることが報告されている。また、「幹細胞」の維持には、DNA損傷後のチェックポイント機構等の細胞周期制御も重要であることも考えられる(図3)。
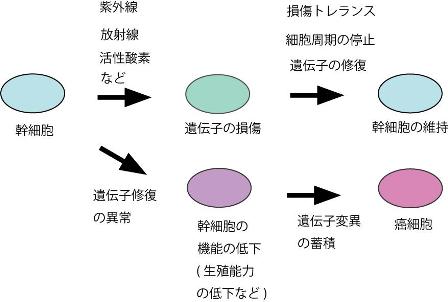
図 3. 幹細胞の維持機構とその破綻による機能低下と発癌
幹細胞の DNAが活性酸素などにより損傷されると、細胞周期が制御され損傷部位が修復される。損傷が残存していても損傷トレランス機構により幹細胞の自律的複製を維持していると考えられる。これらの機構により、幹細胞の数と機能が維持されている。これに対して、DNA修復機構や損傷トレランス機能が低下すると、幹細胞の数および機能低下に伴い、生殖能力および免疫能力等が低下する。遺伝子の変異の蓄積が大きい場合には、幹細胞は癌細胞に変化する可能性もある。
また、近年話題となっている「癌幹細胞」の維持には、「 DNA修復因子」と「細胞周期の制御因子」が一定の役割を果たしていることが報告されている。正常な幹細胞と比較し、どのような点でこの2つの機構に関して差異がみられるか詳細に検討することが、今後の重要な課題である。
7. RAD18によるDNA2重鎖切断損傷の修復促進機構
X線照射などにより、DNA2重鎖が切断される。DNA2重鎖切断損傷は、XRCC4/LigaseIVなどによる非相同末端再結合(NHEJ)またはRAD51などによる組換え機構により修復される。前者は細胞周期がG1期で、後者はG2/M期で機能する。我々は、RAD18がDNA2重鎖切断部位にも集積することを明らかにした 17) 。DNA2重鎖切断部位にはリン酸化ヒストンH2AX、Nbs1、Brca1、53BP1などのタンパクが集積することが知られており、RAD18と共に局在することがわかった。RAD18がG1期でDNA2重鎖切断部位に集積するには、53BP1が必要であることがわかった。精製したRAD18はIn vitroで53BP1タンパクの1268番目のリジン残基をモノユビキチン化する活性を示した。このアミノ酸を変異させた53BP1は、DNA2重鎖切断部位への集積能力が著しく低下しており、またFRAP解析の結果、DNA2重鎖切断部位での安定性が大きく低下していた 17) 。53BP1をモノユビキチン化できない変異RAD18を発現する細胞は、G1期でのX線に対する感受性がみられた。このため、DNA2重鎖切断部位のヒストンH2AXに付加されたポリユビキチン鎖に結合したRAD18は、53BP1をモノユビキチン化することにより53BP1のDNA2重鎖切断部位での安定性を増加することにより、53BP1による非相同末端結合(NHEJ)機構によりDNA2重鎖切断損傷を修復するモデルを提唱した 15) 。
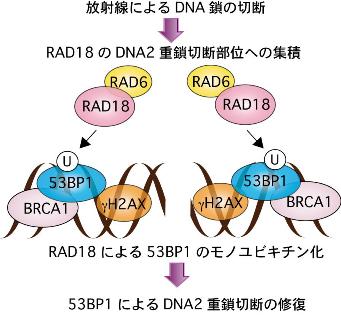
図 4 RAD18によるDNA2重鎖切断損傷の修復促進機構
放射線の照射または抗癌剤の投与により、 DNAが切断されることによりDNA2重鎖切断損傷が形成される。これに応答して切断部位にリン酸化ヒストンH2AX,Nbs1,Brca1または53BP1等の因子が集積することが知られている。RAD18も切断部位に集積し、53BP1の1268番目のリジン残基をモノユビキチン化修飾することにより、53BP1のクロマチンへの結合力が高める。これにより53BP1による非相同末端結合(NHEJ)によるDNA2重鎖切断損傷の修復を促進する。
8.RAD18とがんとの関係
ヒトの初代培養細胞では RAD18の発現量が少ないのに対し、この細胞をSV40ウイルス由来DNAを用いて細胞株(不死化)の状態にすると、RAD18の発現量が増大する。また、多くのヒトがん由来の細胞株でRAD18の発現量が高いことがわかった。マウスの皮膚組織においても、増殖が活発な領域でRAD18の高い発現が観察される。おそらくS期でRAD18の発現が正に制御されていると考えられる 14) 。ただし、マウス野生型細胞とRAD18欠損細胞を比較すると、通常の状態では増殖速度に差はみられない 7) 。RAD18は細胞増殖そのものではなく、複製フォークの進行を円滑に進めるために必要な因子であるためDNA複製時に発現が正に調節されていると推測される。このため、DNA損傷を形成させるタイプの抗がん剤に対する耐性に、RAD18の発現量が影響を及ぼしている可能性が考えられる。多くの方に興味をもっていただけると、うれしく思います。
文献
1) Broomfield, S., Hryciw, T. & Xiao, W. (2001) Mutat. Res., 486, 167-184.
2) Masutani, C., Kusumoto, R., Yamada, A., Dohmae, N., Yokoi, M., Yuasa, M., Araki, M., Iwai, S., Takio, K. & Hanaoka, F. (1999) Nature, 399, 700-704.
3) Matsuda, T., Bebenek, K., Masutani, C., Hanaoka, F. & Kunkel, T. A. (2000) Nature , 404, 1011-1013.
4) Prakash, L. (1981) Mol. Gen. Genet ., 184, 471-478.
5) Hoege, C., Pfander, B., Moldovan, G. L., Pyrowolakis, G. & Jentsch, S. (2001) Nature, 419 135-141.
6) Tateishi, S., Sakuraba Y., Masuyama, S., Inoue, H. & Yamaizumi, M. (2000) Proc. Natl. Acad. Sci. USA, 97, 7927-7932.
7) Tateishi, S., Niwa, H., Miyazaki, J., Fujimoto, S., Inoue, H. & Yamaizumi, M. (2003) Mol. Cell. Biol., 23, 474-481.
8) Watanabe, K., Tateishi, S., Kawasuji, M., Tsurimoto, T., Inoue, H. & Yamaizumi, M. (2004) EMBO J., 23, 3886-3896.
9) Kannouche, P., Broughton, B. C., Volker, M., Hanaoka, F., Mullenders, L. H. & Lehmann, A. R. (2001)Genes Dev., 15, 158-172.
10) Yuasa, M. S., Masutani, C., Hirano, A., Cohn, M. A., Yamaizumi, M., Nakatani, Y. & Hanaoka, F. (2006) Genes Cell,s 11, 731-744.
11) Tsuji, Y., Watanabe, K., Araki, K., Shinohara, M., Yamagata , Y., Tsurimoto, T., Hanaoka, F., Yamamura, K., Yamaizumi, M., and Tateishi, S. (2008) Genes to Cells , 13, 343-354
12) Kannouche, P. L., Wing, J. & Lehmann, A. R. (2004) Mol. Cell , 14, 491-500.
13) Ohmori, H. Friedberg, E. C., Fuchs, R. P., Goodman, M. F., Hanaoka, F., Hinkle, D., Kunkel, T. A., Lawrence, C. W., Livneh, Z., Nohmi, T., Prakash, L., Prakash, S., Todo, T., Walker, G. C., Wang, Z., Woodgate, R. (2001) Mol. Cell, 8, 7-8.
14) Sun, J., Yomogida, K., Sakao, S., Yamamoto, H., Yoshida, K., Watanabe, K., Morita, T., Araki, K., Yamamura, K. and Tateishi, S. (2008) Mech. Dev. In Press
Watanabe, K., Iwabuchi, K., Sun, J., Tsuji, Y., Tani, T., Tokunaga, K., Date, T., Hashimoto, M., Yamaizumi, M. and * Tateishi, S. (2009) Nucleic Acids Res. doi: 10.1093/nar/gkp082








